Warm Up with RNA-seq data
Warm Up with RNA-seq data
Welcome!
Please open the Vmware machine and log in using the super secret password.
TRY
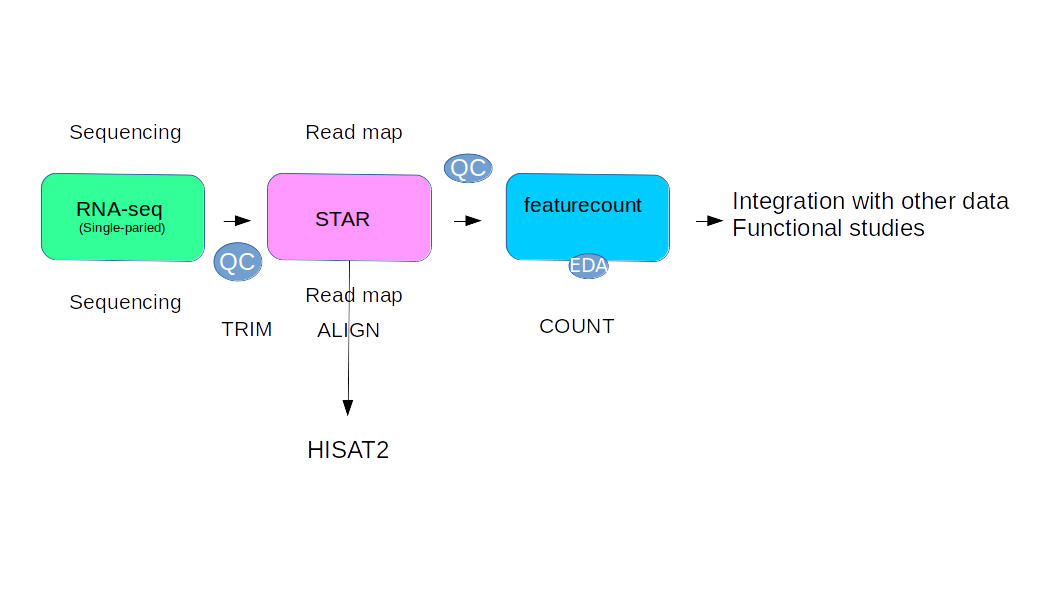
Here we make step by step the rnaseq data pipeline using STAR and htseqcount for calclulare the number of reads correspond for each gene. We use a tutorial publich on this site LINK
The experiment allowed the effect of bad replicates to be assessed as well as providing guidelines for the number of replicates required for differential gene expression analysis and the most appropriate statistical tools
We use this procedure:
[Data_tutorial ](https://www.dropbox.com/s/k3v0y7v9kwj38x9/Star_Practical1.tar.gz?dl=0)
To download the fastq files and simultaneously sort them into appropriate folders, we are going to use two text files that contain sample information, i.e. (i) the information about where the raw reads can be downloaded from (the actual URLs for every single sample) and (ii) the mapping of the accession numbers to the file names.
These text files will be downloaded using the wget command with the following syntax:
wget -O <output name into which the link's content will be downloaded> "<URL>"
The links to these two files of interest were obtained by visiting www.ebi.ac.uk/ena and searching for the project number “ERP004763”.
# download the table of samples to the server in text format
wget -O samples_at_ENA.txt "https://www.ebi.ac.uk/ena/data/warehouse/filereport?accession=PRJEB5348&result=read_run&fields=study_accession,sample_accession,secondary_sample_accession,experiment_accession,run_accession,tax_id,scientific_name,instrument_model,library_layout,fastq_ftp,fastq_galaxy,submitted_ftp,submitted_galaxy,sra_ftp,sra_galaxy,cram_index_ftp,cram_index_galaxy&download=txt"
# a file provided by the authors of the study outside the actual repository
wget -O sample_mapping.txt "https://ndownloader.figshare.com/files/2194841"
The files should have the following content:
$ head sample_mapping.txt
RunAccession Lane Sample BiolRep
ERR458493 1 WT 1
ERR458494 2 WT 1
ERR458495 3 WT 1
ERR458496 4 WT 1
ERR458497 5 WT 1
ERR458498 6 WT 1
ERR458499 7 WT 1
ERR458500 1 SNF2 1
ERR458501 2 SNF2 1
$ cut -f11 samples_at_ENA.txt | head
fastq_galaxy
ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458493/ERR458493.fastq.gz
ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458494/ERR458494.fastq.gz
ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458495/ERR458495.fastq.gz
ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458496/ERR458496.fastq.gz
ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458497/ERR458497.fastq.gz
ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458498/ERR458498.fastq.gz
ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458499/ERR458499.fastq.gz
ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458500/ERR458500.fastq.gz
ftp.sra.ebi.ac.uk/vol1/fastq/ERR458/ERR458501/ERR458501.fastq.gz
To download some files of interest, you can proceed as follows:
-
find out which RunAccession numbers belong to the WT and SNF2 samples of BiolRep #1
# show all columns ($0 is equivalent to 'all columns') of all rows where the 4th column equals 1 awk '$4 == 1 {print $0}' sample_mapping.txt -
download an individual sample for which you know the acession number
# extract the 11th column (which contains the actual URL) from the row where the 5th column corresponds to the accession number of interest (ERR458493) and then supply that link to wget using xargs awk -F "\t" '$5 == "ERR458493" {print $11}' samples_at_ENA.txt | xargs wget -
either do this 6 more times individually supplying every accession number of interest or write a for-loop that makes use of the fact that all WT samples of interest seem to start with
ERR45849(493 through 499) and all SNF2 samples of interest start withERR45850(500 through 506). Tip: see what just typingseq 3 9results in.- for-loop for WT samples
# create a folder to downlaod the raw reads to mkdir WT_1 cd WT_1 # write the loop to repeat the wget-based command from above for every WT (repl. 1) sample of interest for i in `seq 3 9` do SAMPLE=ERR45849${i} egrep ${SAMPLE} ../samples_at_ENA.txt | cut -f11 | xargs wget done- for-loop for SNF2 samples
cd ../ mkdir SNF2_1 cd SNF2_1 for i in `seq 0 6` do SAMPLE=ERR45850${i} egrep ${SAMPLE} ../samples_at_ENA.txt | cut -f11 | xargs wget done
You should now see the fastq.gz files for the first biological replicates of both WT and SNF2 (7 files each):
cd ../
ls WT_1
ls SNF2_1
This is a rather ad-hoc way of doing this since we still use the observation about the accession number patterns for the samples belonging to WT_repl1 and SNF2_repl1. To see a more stringent and less error-prone way, check out the README of how we downloaded ALL the samples: ~/mat/precomputed/rawReads_yeast_Gierlinski/README_sampleDownload .
THIS IS JUST FOR DEMONSTRATION, DO NOT DOWNLOAD ALL THE SAMPLES!
QC of RAW READS
We will make a folder where we are going to keep track of all the different QC steps, so let’s start by running FastQC.
Note that the following file paths assume that you’re within the folder class, which comes right after your home directory (~/class). Type pwd if you aren’t sure where you are.
Install enviroment:
conda create -n Test2
conda activate Test2
conda install fastqc -y
conda install multiqc -y
conda install STAR -y
conda install samtools -y
conda install qorts -y
mkdir qc_raw_reads
for SAMPLE in WT_1 SNF2_1
do
mkdir qc_raw_reads/${SAMPLE}
fastqc ${SAMPLE}/*fastq.gz \
-o qc_raw_reads/${SAMPLE}
done
Check the FastQC output - there should be 7 html files and new folders per sample:
ls qc_raw_read/WT_1
ls qc_raw_read/SNF2_1
Summarize those into one report using MultiQC:
cd qc_raw_reads/
multiqc . --dirs --filename multiQC_rawReads
Send the results to yourself:
echo "FastQC results for WT_1 and SNF2_1" | mailx -s "FastQC results" -a "multiQC_rawReads.html" YOUR_EMAIL@wherever.com
or download them to your computer:
# the following command assumes you're logged OUT of the server and somewhere sensible on your own machine, e.g. in ~/Downloads
scp <your login name>@<server IP>:/home/<your login name>/class/qc_raw_reads/multiQC_rawReads.html .
The report should show you that:
- the two samples have very similar GC contents, numbers of reads and duplication rates
- the GC content for
SNF2_rep1andWT_rep1, however, are distinct, reflecting the library-specific biases in regard to GC - one technical replicate of
WT_rep1has a slightly elevated adapter content
For an example MultiQC result (following FastQC on 112 fastq files), check this out.
ALIGN DATA
mkdir reference
cd reference
wget -r -nd ftp://ftp.ensembl.org/pub/release-96/fasta/saccharomyces_cerevisiae/dna/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa.gz
wget -r -nd ftp://ftp.ensembl.org/pub/release-96/gtf/saccharomyces_cerevisiae/Saccharomyces_cerevisiae.R64-1-1.96.gtf.gz
How extract these files?
ALign on reference
So we can write only single code or we can align more quickly using a script. Here I show you a way to do the analisys using a bash script. Here we start creating the reference on STAR
STAR --runMode genomeGenerate --genomeDir reference/ --genomeFastaFiles reference/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa --sjdbGTFfile reference/Saccharomyces_cerevisiae.R64-1-1.96.gtf --sjdbOverhang 100
Then we start to allign all the data using this code:
for SAMPLE in WT_1 SNF2_1
do
STAR --runMode alignReads --genomeDir reference/ --readFilesIn ${SAMPLE}/*.fastq.gz --readFilesCommand zcat --outStd Log --outSAMunmapped Within --outSAMtype BAM SortedByCoordinate --runThreadN 1 --outFileNamePrefix bamout/${SAMPLE}
done
miti=(`find WT_1/ -name "*.gz"`)
for i in "${miti[@]}";
do
echo $i,name=$(echo $i|awk -F"/" '{print $2}'|sed 's/.fastq.gz//g');
STAR --runMode alignReads --genomeDir reference/ --readFilesIn $i --readFilesCommand zcat --outStd Log --outSAMunmapped Within --outSAMtype BAM SortedByCoordinate --runThreadN 1 --outFileNamePrefix bamout/$name
done
miti=(`find SNF2_1/ -name "*.gz"`)
for i in "${miti[@]}";
do
echo $i,name=$(echo $i|awk -F"/" '{print $2}'|sed 's/.fastq.gz//g');
STAR --runMode alignReads --genomeDir reference/ --readFilesIn $i --readFilesCommand zcat --outStd Log --outSAMunmapped Within --outSAMtype BAM SortedByCoordinate --runThreadN 1 --outFileNamePrefix bamout/$name
done
We can Merge the log results:
### STAR log files
Just need to be copied from the alignment folder:
for SAMPLE in SNF2_1 WT_1
do
for i in baout/${SAMPLE}/*Log*final.out
do
echo "cp " $i ${SAMPLE}"/STAR_log.final.out"
done
done
QoRTs
As an alternative to MultiQC, QoRTs can be used to summarize multiple QC checks in one file. Unfortunately in this machine not work but at least you know how to run
# to call up the help manual for the QC module of QoRTs:
java -Xmx4g -jar ~/mat/software/qorts.jar QC man
QoRTs produces a lot of individual files if run in QC mode.
for SAMPLE in WT_1 SNF2_1
do
java -Xmx4g -jar ~/mat/software/qorts.jar QC \
--singleEnded \
--generatePdfReport \
bam_files/${SAMPLE}/*.bam ${REF_DIR}/sacCer3.gtf $SAMPLE
done
The inclusion of QoRTs doesn’t fully work yet.
Bonus
In order to improve the alignment results for the yeast samples, we re-run the alignment for several samples. The most important parameters that we tuned were related to the intron sizes.
To determine suitable numbers for IntronMin and IntronMax parameters,
we downloaded a bed file for the yeast introns from UCSC table browser (details at biostars) We found from 1 to 3000. We can re-run STAR with the new parameeter
for SAMPLE in WT_1 SNF2_1
do
FILES=`ls -m ${SAMPLE}/*fastq.gz | sed 's/ //g'`
FILES=`echo $FILES | sed 's/ //g'`
echo "Aligning files for ${SAMPLE}, files:"
echo $FILES
$runSTAR --genomeDir reference/ --readFilesIn $FILES --readFilesCommand zcat --outFileNamePrefix ${SAMPLE}_ --outFilterMultimapNmax 1 --outSAMtype BAM SortedByCoordinate --runThreadN 1 --twopassMode Basic --alignIntronMin 1 --alignIntronMax 3000
done