RNA-Seq data analysis
RNA-seq data analyss with different approachs.

Introduction
RNA-Seq (RNA sequencing ) also called whole transcriptome sequncing use next-generation sequeincing (NGS) to reveal the presence and quantity of RNA in a biolgical sample at a given moment.
Practical 1
In this session we want to perform some differential expressin from two condition as example (Normal vs tumor RNA-seq). This data use for this tutorial are pubblicaly avaible.
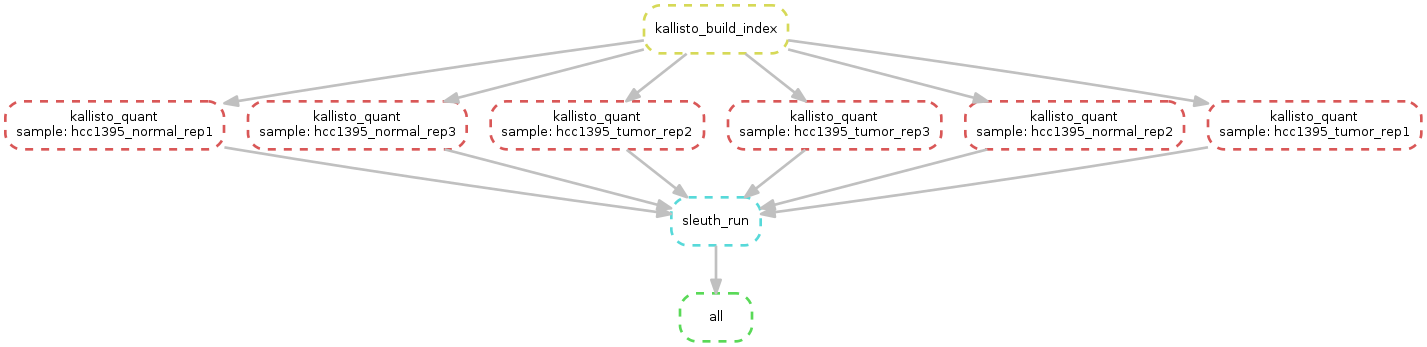
Please dwnlad the material here: For count Pipeline TEST Please follow this command
cd ~/Desktop/
tar xvf TUTORIAL_RNA_snakemake.tar.gz
cd TUTORIAL
conda env create -f snakemake_differential.yml
Please launch the analysis:
snakemake -s Snakefile.Kallisto --configfile database3.yml --use-conda
Please Conect and see this tutorial on live sleuth
[youtube](https://www.youtube.com/watch?v=KEn0CMYk6Wo)
PRACTICAL COUNTS DATA
In the previous example we use the new way to do RNA-seq analisys using pseudoma p approach. This method are recently reported (Kallisto and Salmon). Another way to do this type analisys was performed using counts. Normally you can allign with programs like STAR and count gene and isoforms using HTseq-count or feature-count. Here we are some examples of working on R oon Counts.
conda create -n DESEQ
source activate DESEQ
conda install -y r-mixomics bioconductor-htsfilter bioconductor-deseq2
R
library(DESeq2)
library(RColorBrewer)
library(mixOmics)
library(HTSFilter)
directory <- "RNAseq_data/count_table_files/"
dir(directory)
*Exercise 3.1* Read the files:
rawCountTable <- read.table(paste0(directory,"count_table.tsv"), header=TRUE,
row.names=1)
sampleInfo <- read.table(paste0(directory,"pasilla_design.txt"), header=TRUE,
row.names=1)
*Exercise 3.2* Have a look at the count data:
head(rawCountTable)
nrow(rawCountTable)
*Exercise 3.3* Have a look at the sample information and order the count table
in the same way that the sample information:
sampleInfo
rawCountTable <- rawCountTable[,match(rownames(sampleInfo),
colnames(rawCountTable))]
head(rawCountTable)
*Exercise 3.4* Create the 'condition' column
sampleInfo$condition <- substr(rownames(sampleInfo), 1,
nchar(rownames(sampleInfo))-1)
sampleInfo$condition[sampleInfo$condition=="untreated"] <- "control"
sampleInfo$condition <- factor(sampleInfo$condition)
sampleInfo
*Exercise 3.5* Create a 'DESeqDataSet' object
ddsFull <- DESeqDataSetFromMatrix(as.matrix(rawCountTable), sampleInfo,
formula(~condition))
ddsFull
# 3.2. Starting from separate files
*Exercise 3.6* List all files in directory
directory <- "RNAseq_data/separate_files/"
sampleFiles <- list.files(directory)
sampleFiles
*Exercise 3.7* Create an object with file informations
keptFiles <- sampleFiles[-1]
sampleName <- sapply(keptFiles, function(afile)
substr(afile, 1, nchar(afile)-6))
condition<- sapply(keptFiles, function(afile)
substr(afile, 1, nchar(afile)-7))
fileInfo <- data.frame(sampleName = sampleName, sampleFiles = keptFiles,
condition = condition)
rownames(fileInfo) <- NULL
fileInfo
*Exercise 3.8* Construct a 'DESeqDataSet' object
ddsHTSeq <- DESeqDataSetFromHTSeqCount(fileInfo, directory, formula(~condition))
ddsHTSeq
# 3.3. Preparing the data object for the analysis of interest
*Exercise 3.9* Select the subset of paire-end samples
dds <- subset(ddsFull, select=colData(ddsFull)$type=="paired-end")
dds
colData(dds)
# 3.4 Data exploration and quality assessment
*Exercise 3.10* Extract pseudo-counts (*ie* $\log_2(K+1)$)
pseudoCounts <- log2(counts(dds)+1)
head(pseudoCounts)
*Exercise 3.11* Histogram for pseudo-counts (sample ```treated2```)
```{r histoPseudoCounts}
hist(pseudoCounts[,"treated2"])
*Exercise 3.12* Boxplot for pseudo-counts
boxplot(pseudoCounts, col="gray")
*Exercise 3.13* MA-plots between control or treated samples
par(mfrow=c(1,2))
## treated2 vs treated3
# A values
avalues <- (pseudoCounts[,1] + pseudoCounts[,2])/2
# M values
mvalues <- (pseudoCounts[,1] - pseudoCounts[,2])
plot(avalues, mvalues, xlab="A", ylab="M", pch=19, main="treated")
abline(h=0, col="red")
## untreated3 vs untreated4
# A values
avalues <- (pseudoCounts[,3] + pseudoCounts[,4])/2
# M values
mvalues <- (pseudoCounts[,3] - pseudoCounts[,4])
plot(avalues, mvalues, xlab="A", ylab="M", pch=19, main="control")
abline(h=0, col="red")
*Exercise 3.14* PCA for pseudo-counts
vsd <- varianceStabilizingTransformation(dds)
vsd
plotPCA(vsd)
*Exercise 3.15* heatmap for pseudo-counts (using ```mixOmics``` package)
sampleDists <- as.matrix(dist(t(pseudoCounts)))
sampleDists
cimColor <- colorRampPalette(rev(brewer.pal(9, "Blues")))(16)
cim(sampleDists, col=cimColor, symkey=FALSE)
# 3.5. Differential expression analysis
*Exercise 3.16* Run the DESeq2 analysis
dds <- DESeq(dds)
dds
# 3.6. Inspecting the results
*Exercise 3.17* Extract the results
res <- results(dds)
res
*Exercise 3.18* Obtain information on the meaning of the columns
mcols(res)
*Exercise 3.19* Count the number of significant genes at level 1%
sum(res$padj < 0.01, na.rm=TRUE)
*Exercise 3.20* Extract significant genes and sort them by the strongest down
regulation
sigDownReg <- res[!is.na(res$padj), ]
sigDownReg <- sigDownReg[sigDownReg$padj < 0.01, ]
sigDownReg <- sigDownReg[order(sigDownReg$log2FoldChange),]
sigDownReg
*Exercise 3.21* Extract significant genes and sort them by the strongest up
regulation
sigUpReg <- res[!is.na(res$padj), ]
sigUpReg <- sigUpReg[sigUpReg$padj < 0.01, ]
sigUpReg <- sigUpReg[order(sigUpReg$log2FoldChange, decreasing=TRUE),]
*Exercise 3.22* Create permanent storage of results
write.csv(sigDownReg, file="sigDownReg-deseq.csv")
write.csv(sigUpReg, file="sigUpReg-deseq.csv")
# 3.7 Diagnostic plot for multiple testing
*Exercise 3.23* Plot a histogram of unadjusted p-values after filtering
hist(res$pvalue, breaks=50)
# 3.8 Interpreting the DE analysis results
*Exercise 3.24* Create a MA plot showing differentially expressed genes
plotMA(res, alpha=0.01)
*Exercise 3.25* Create a Volcano plot
volcanoData <- cbind(res$log2FoldChange, -log10(res$padj))
volcanoData <- na.omit(volcanoData)
colnames(volcanoData) <- c("logFC", "negLogPval")
head(volcanoData)
plot(volcanoData, pch=19, cex=0.5)
*Exercise 3.26* Transform the normalized counts for variance stabilization
vsnd <- varianceStabilizingTransformation(dds, blind=FALSE)
vsnd
*Exercise 3.27* Extract the transformed data
head(assay(vsnd), 10)
selY <- assay(vsnd)[!is.na(res$pval), ]
selY <- selY[res$pval[!is.na(res$pval)] < 0.01,]
cimColor <- colorRampPalette(rev(brewer.pal(9, "Blues")))(255)[255:1]
cim(t(selY), col=cimColor, symkey=FALSE)
KALLISTO PIPELINE USING NEXTFLOW
Here antoher way to do the analysis. Please familiarize with the results
cd /home/bioinfo2/Data/kallisto-nf
sudo nextflow run cbcrg/kallisto-nf -with-docker
OPTIONAL PRATICAL
Please follow this tutorial [link] (http://www.nathalievilla.org/doc/html/solution_edgeR-tomato.html#where-to-start-installation-and-alike) Pratical rnaseq data using tomato data