General installation for Fusion detection tutorial
Comand line preparation enviroment for our tutorial.
General installation for Fusion detection tutorial
Links
Introduction
In this part of the course, we will present the main steps that are commonly used to process and to analyze RNA-seq cancer sequencing data for detect fusion transcript genes. This workshop will show you how to launch individual steps of a complete RNA-seq pipeline for detect fusion with some software. All the softwares are already installed on your virtual machine. If you need assistance please let me know.
Software requirements
Fusion detection softwares have different requirement and linux machine are normaly used for this purpose. Exist however the possibility to use some virtual container like docker that allow to use this software also in another operative system (OS,Windows).
We concentrate our tutorial only this software :
- STAR-FUSION [https://github.com/STAR-Fusion/STAR-Fusion/wiki]
- JAFFA [https://github.com/Oshlack/JAFFA/wiki]
- Defuse [https://bitbucket.org/dranew/defuse]
- Fusioncather [https://github.com/ndaniel/fusioncatcher]
Defuse and Fusioncather are not avaible in your virtual machines. Another important program fusioncather are unfortunately not describe with example in this tutorial for time and resource avaible. The Ensemble of many tools for detection fusion transcript are the best solution avaible. In the next months more information on the other programs will be pubblish.
Original Setup
The initial structure of your folders should look like this:
├── RAW
│ ├── BT474-demo_1.fastq.gz
│ ├── BT474-demo_2.fastq.gz
│ ├── MCF7-demo_1.fastq.gz
│ └── MCF7-demo_2.fastq.gz
├── README.md
├── Results
│ ├── HCC1395
│ │ ├── defuse.tar.gz
│ │ └── starfusion.tar.gz
│ ├── JAFFA
│ │ ├── BT474_MCF7
│ │ │ └── ASSEMBLY
│ │ │ ├── checks
│ │ │ ├── commandlog.txt
│ │ │ ├── jaffa_results.csv
│ │ │ └── jaffa_results.fasta
│ │ ├── BT474_MCF7.tar.gz
│ │ └── Simulazione_50x.tar.gz
│ └── STAR-FUSION
│ ├── Chimeric.out.junction
│ ├── Sim_50x
│ │ ├── star-fusion.fusion_candidates.final
│ │ ├── star-fusion.fusion_candidates.final.abridged.FFPM
│ │ ├── star-fusion.fusion_candidates.preliminary
│ │ └── star.ok
│ └── test_kickstart
│ ├── star-fusion.fusion_candidates.final
│ └── star-fusion.fusion_candidates.preliminary
└── scripts
├── _run_starfusion.sh
└── _script.jaffa.sh
10 directories, 22 file
If you don’t have this directory please download this and execute this commmand:
git clone https://bitbucket.org/biomauro/tutorial-fusion-transcript-2017
Here you have the basic structure of the data and the way to download all the data you need to start this tutorial.
Sample Information
For this tutorial we use 3 samples:
- BTF-474 (SRA accession SRR925695)filter dataset
-
MCF-7 (SRA accession SRR925723)filter dataset
-
testing Chimeric.out.junction of the BTF-474 from STAR-FUSION repository link
-
Fusion_simulation_50x (create in house each fusion have 50X coverage)
- Fusion of HCC1395 from Andrew McPherson tutorial
The first two samples are used only for jaffa tutorial. This demo should only take a few minutes, whereas a full dataset may take hours. The reads are 76bp paired end, which means we can show all three modes of JAFFA: assembly, direct and hybrid.
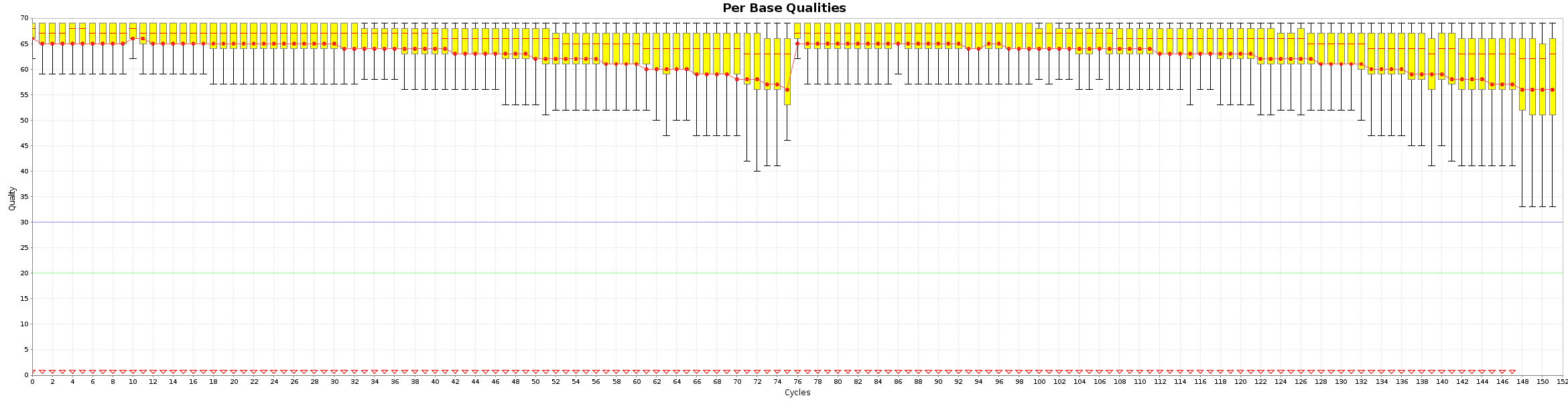
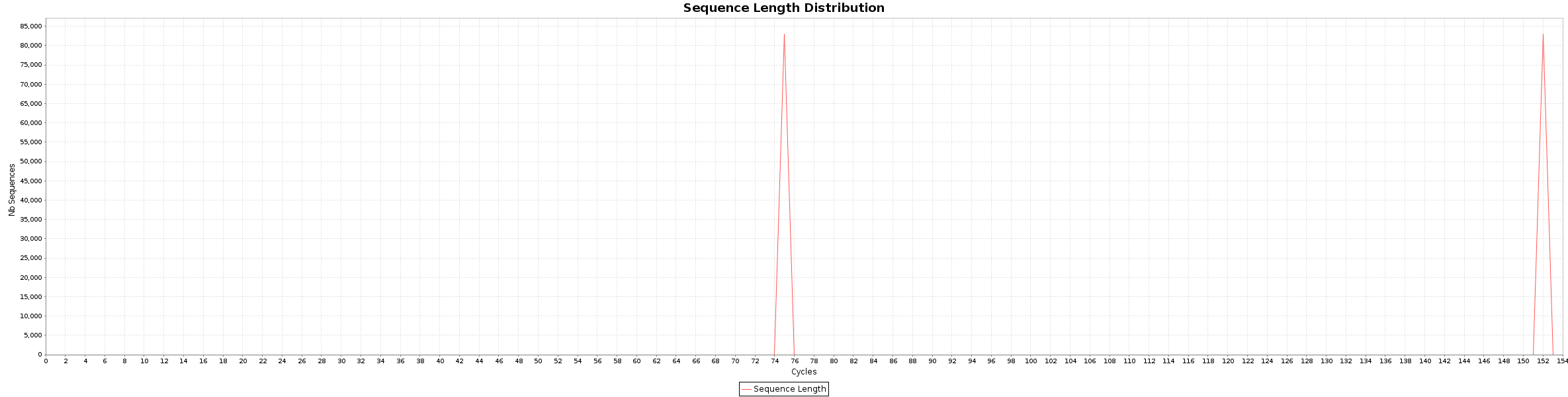
The Fusion_simulation_50x library is a RNA-seq data from Normal Sample (bodymap) where we add some fusion. The library are created as pair-end 2x50 bp.
$ java -jar /opt/bvatools-1.6/bvatools-1.6-full.jar readsqc --read1 ../RAW/reads_50x_R1.fq.gz --read2 ../RAW/reads_50x_R2.fq.gz
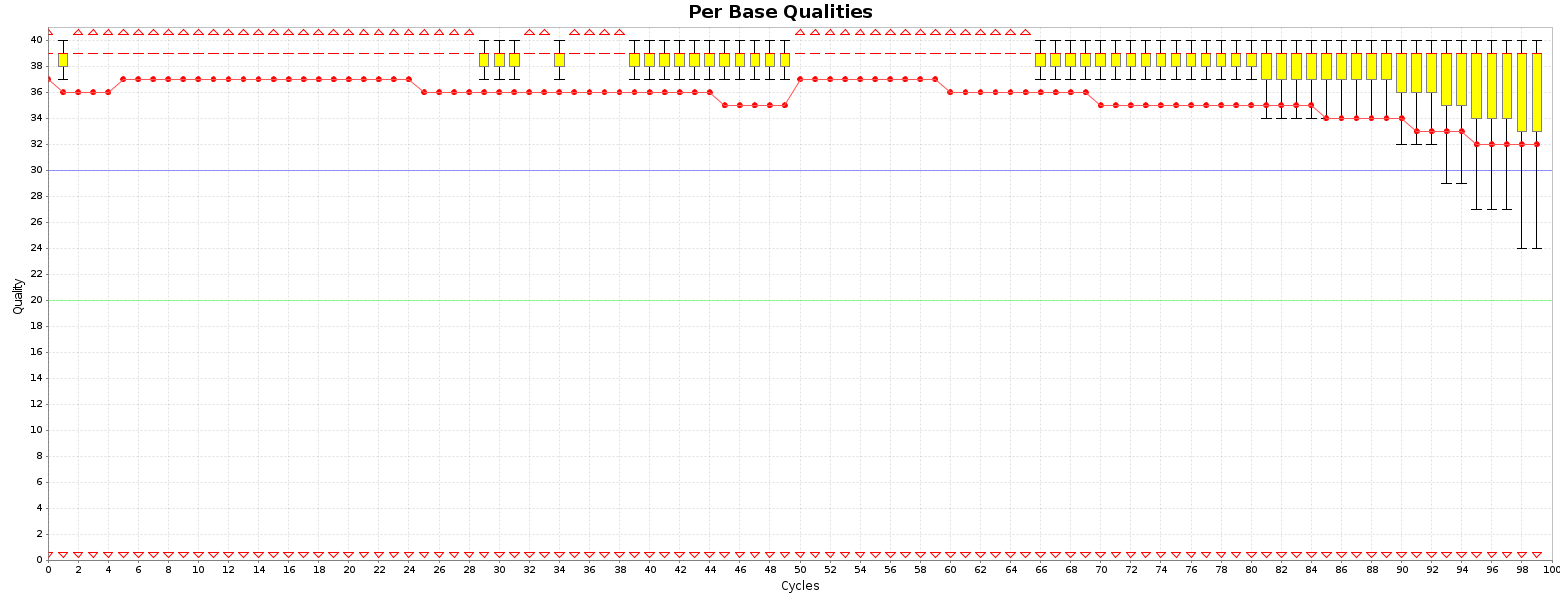
We run this simulation library to this softwares:
- JAFFA (Assembly)
- Defuse (last version 2017)
- STAR-FUSION (v1.1.0)
This datasets can be use to become familiar to the ouput of this tools.
Sample HCC1395 derived from tutorial develope by Andrew McPherson on cbw.