RNA-seq Introduction
RNA-seq Introduction
Introduction
The original goal of RNA sequencing was to identify which genomic loci are expressed in a cell (population)at a given time over the entire expression range, i.e., to offer a superior alternative to cDNA microarrays. Indeed, RNA-seq was shown to detect lowly expressed transcripts while suffering from strongly reducedfalse positive rates in comparison to microarray based expression quantification (Illumina, 2011; Nookaewet al., 2012; Zhao et al., 2014)∗. Since RNA-seq does not rely on a pre-specified selection of cDNA probes,there are numerous additional applications of RNA-seq that go beyond the counting of expressed transcriptsof known genes, such as the detection and quantification of non-genic transcripts, splice isoforms, noveltranscripts and protein-RNA interaction sites. The detection of gene expression changes (i.e., mRNA levels)between different cell populations and/or experimental conditions remains the most common application of RNA-seq. Nevertheless, unlike for genome-based high-throughput sequencing approaches, the RNA-seq field lacks widely accepted and adopted standards and is only slowly coming to terms about best practices fordifferential gene expression analysis amid a myriad of available software.
Workflow of a differential gene expression
Sequencing (biochemistry)(a) RNA extraction(b) Library preparation (including mRNA enrichment)(c) Sequencing2.Bioinformatics(a) Processing of sequencing reads (including alignment)(b) Estimation of individual gene expression levels(c) Normalization(d) Identification of differentially expressed (DE) genes
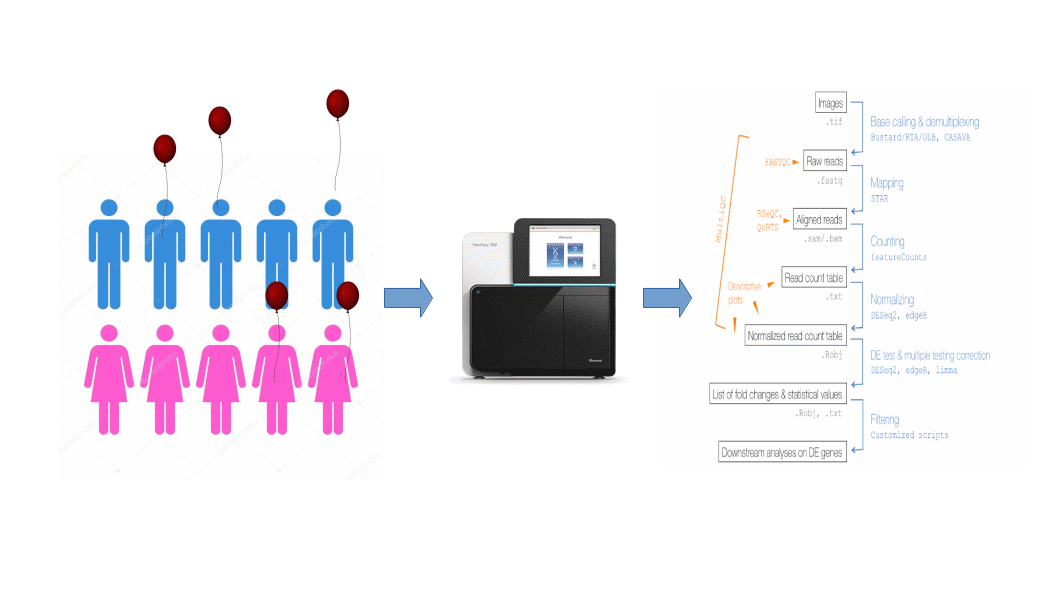
RNAseq can be roughly divided into two “types”:
- **Reference genome-based** - an assembled genome exists for a species for which an RNAseq experiment is performed. It allows reads to be aligned against the reference genome and significantly improves our ability to reconstruct transcripts. This category would obviously include humans and most model organisms but excludes the majority of truly biologically interesting species (e.g., Hyacinth macaw);
- **Reference genome-free** - no genome assembly for the species of interest is available. In this case one would need to assemble the reads into transcripts using de novo approaches. This type of RNAseq is as much of an art as well as science because assembly is heavily parameter-dependent and difficult to do well

Replicates: Biological or Technical and how many?
An RNAseq experiment without a sufficient number of replicates will be a waste of money. Replicates are essential to be able to correct for variation due to differences within/among organisms, cells, sequencing machines, library preparation protocols and numerous other potential factors. There are two types of replicates (as described by Dündar:2015):
- **Technical replicates** can be defined as different library preparations from the same RNA sample. They should account for batch effects from the library preparation such as reverse transcription and PCR amplification. To avoid possible lane effects (e.g., differences in the sample loading, cluster amplification, and efficiency of the sequencing reaction), it is good practice to multiplex the same sample over different lanes of the same flowcell. In most cases, technical variability introduced by the sequencing protocol is quite low and well controlled.
- **Biological replicates.** There is an on-going debate over what kinds of samples represent true biological replicates. Obviously, the variability between different samples will be greater between RNA extracted from two unrelated humans than between RNA extracted from two different batches of the same cell line. In the latter case, most of the variation that will eventually be detected was probably introduced by the experimenter (e.g., slightly differing media and plating conditions). Nevertheless, this is variation the researcher is typically not interested in assessing, therefore the ENCODE consortium defines biological replicates as RNA from an independent growth of cells/tissue (ENCODE 2011).
The number of replicates should be as high as practically possible. Most RNAseq experiments include three replicates and some have as many as 12 (see Schurch et al. 2015). Unfortunately we have many time 2 versus 2 o 2 vs 5.
PRACTICAL DISTRIBUTION
We Perform this Practical:
1. RNA-seq count with Hisat2 and Star2
2. Salmon quantification
3. Kallisto quantification
4. Diffential using DESEq2 and airway package
5. Differential using edger