Bioinformatics approach examples
We have many way to write our pipelines. Each approach have some problems. We need to use take in mind what we need to do.

Introduction
In Unix-like computer operating systems, a pipeline is a sequence of processes chained together by their standard streams, so that the output of each process (stdout) feeds directly as input (stdin) to the next one. The concept of pipelines was championed by Douglas McIlroy at Unix’s ancestral home of Bell Labs, during the development of Unix, shaping its toolbox philosophy.[1][2] It is named by analogy to a physical pipeline.
The standard shell syntax for pipelines is to list multiple commands, separated by vertical bars (“pipes” in common Unix verbiage). For example, to list files in the current directory (ls), retain only the lines of ls output containing the string “key” (grep), and view the result in a scrolling page (less), a user types the following into the command line of a terminal:
ls -l | grep key | less
| “ls -l” produces a process, the output (stdout) of which is piped to the input (stdin) of the process for “grep key”; and likewise for the process for “less”. Each process takes input from the previous process and produces output for the next process via standard streams. Each “ | ” tells the shell to connect the standard output of the command on the left to the standard input of the command on the right by an inter-process communication mechanism called an (anonymous) pipe, implemented in the operating system. Pipes are unidirectional; data flows through the pipeline from left to right. |
process1 | process2 | process3
In general a pipeline on bionformatics refers to a prescribed set of processing steps needed to transfer form raw data into something interpretable. This could be for instance, a series of scripts or programs. This is an example of pipeeline for germline calling process encode by GATK GATK
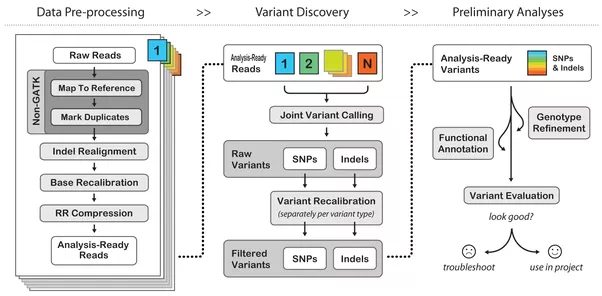
The GATK Best Practices provide step-by-step recommendations for performing variant discovery analysis in high-throughput sequencing (HTS) data. There are several different GATK Best Practices workflows tailored to particular applications depending on the type of variation of interest and the technology employed. The Best Practices documentation attempts to describe in detail the key principles of the processing and analysis steps required to go from raw reads coming off the sequencing machine, all the way to an appropriately filtered variant callset that can be used in downstream analyses. Wherever we can, we try to provide guidance regarding experimental design, quality control (QC) and pipeline implementation options, but please understand that those are dependent on many factors including sequencing technology and the hardware infrastructure that are at your disposal, so you may need to adapt our recommendations to your specific situation.
PRACTICAL
High-throughput bioinformatic analyses increasingly rely on pipeline frameworks to process sequence and metadata. Modern implementations of these frameworks differ on three key dimensions: using an implicit or explicit syntax, using a configuration, convention or class-based design paradigm and offering a command line or workbench interface. Exist many framework for perform our analisys. Here we want to describe 4 methods for perform our analisys.
MANUAL BASH
In this way we write each step on command line and we wait the end for start the new command.
Here we approach the alignment and all the steps need for prepare bam file.
Please familiarize wih this commands:
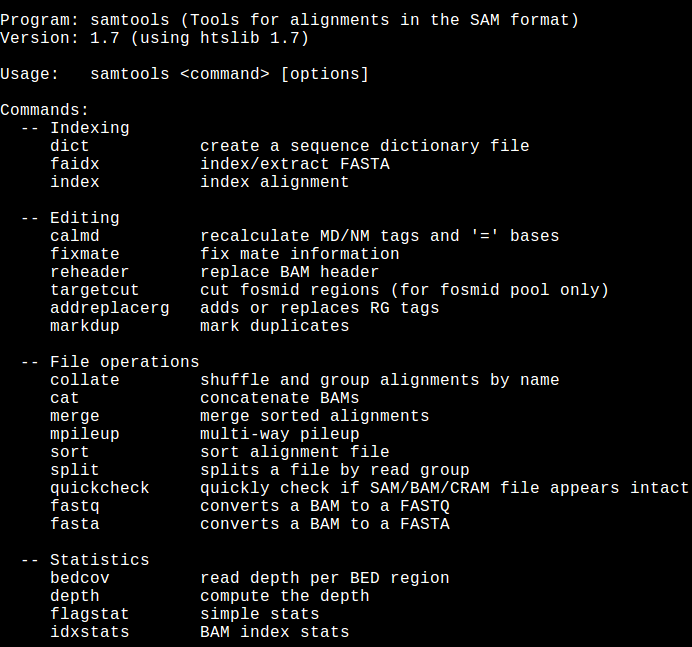
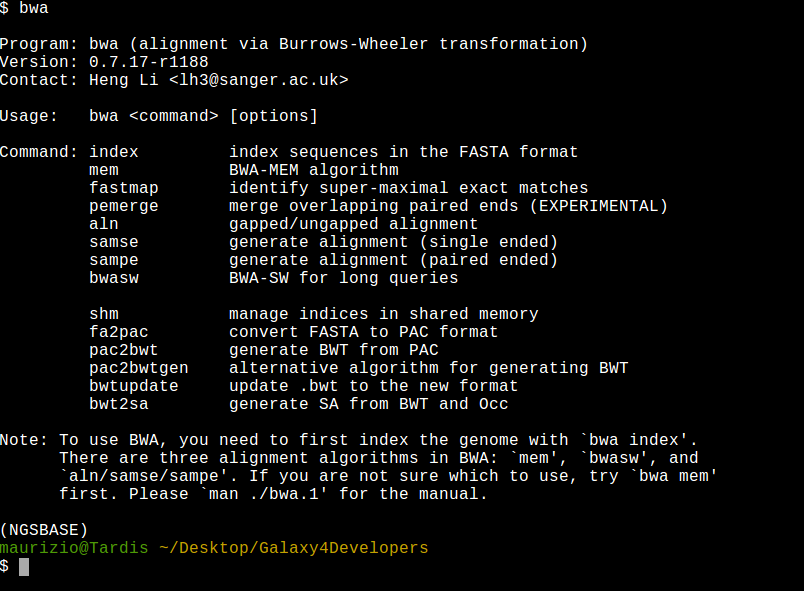
cd ~/Desktop/
#download the data
git clone https://github.com/bioinfo-dirty-jobs/Data.git
# create directory for the execution of programs
mkdir ANALISI1
cd ANALISI1
cp -r ~/Desktop/Data/PIPELINES/* .
#activate enviroment wih all the software previous installed
source activate NGSBASE
When you have finish this part we need to write the commands:
Prepare reference
samtools faidx reference/human_g1k_v37_MT.fasta
picard CreateSequenceDictionary R=reference/human_g1k_v37_MT.fasta o=reference/human_g1k_v37_MT.dict
bwa index reference/human_g1k_v37_MT.fasta
Allign the sample
bwa mem -M -R '@RG\tID:ind1\tSM:ind1\tLB:ind1\tPU:ind1\tPL:Illumina' reference/human_g1k_v37_MT.fasta fastq/ind1_1.fastq.gz fastq/ind1_2.fastq.gz | samblaster -M | samtools fixmate - - | samtools sort -O bam -o bam/ind1.rmdup.sorted.bam -
bwa mem -M -R '@RG\tID:ind2\tSM:ind2\tLB:ind2\tPU:ind2\tPL:Illumina' reference/human_g1k_v37_MT.fasta fastq/ind2_1.fastq.gz fastq/ind2_2.fastq.gz | samblaster -M | samtools fixmate - - | samtools sort -O bam -o bam/ind2.rmdup.sorted.bam -
Prepare statistics file
samtools index bam/ind2.rmdup.sorted.bam
samtools stats bam/ind2.rmdup.sorted.bam | grep ^SN | cut -f 2- > stats/ind2.rmdup.sorted.bam.stats
samtools index bam/ind1.rmdup.sorted.bam
samtools stats bam/ind1.rmdup.sorted.bam | grep ^SN | cut -f 2- > stats/ind1.rmdup.sorted.bam.stats #### Joint call variant for both samples
freebayes -f reference/human_g1k_v37_MT.fasta bam/ind1.rmdup.sorted.bam bam/ind2.rmdup.sorted.bam > vcf/joint.raw.vcf
Please be familiar with the input files we talk yesterday
SCRIPT BASH
You can organize your single step in signle file that run all the pipeline.
This is what we do:
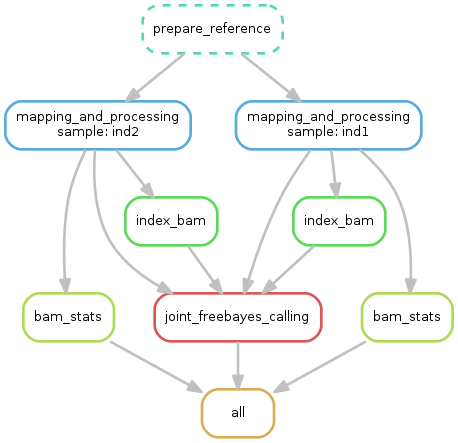
cat example.sh
sh example.sh
RUN USING SNAKEMAKE
You can run all the pipeline uing snakemake link. The Snakemake workflow management system is a tool to create reproducible and scalable data analyses. Workflows are described via a human readable, Python based language. They can be seamlessly scaled to server, cluster, grid and cloud environments, without the need to modify the workflow definition. Finally, Snakemake workflows can entail a description of required software, which will be automatically deployed to any execution environment.
cat example_snakemake
snakemake -s example_snakefile
RUN USING NEXTFLOW
You can run all the pipeline uing nextflow link. Nextflow enables scalable and reproducible scientific workflows using software containers. It allows the adaptation of pipelines written in the most common scripting languages. Its fluent DSL simplifies the implementation and the deployment of complex parallel and reactive workflows on clouds and clusters.
cat Analisi.nf
nextflow run Analisi.nf
REFERENCE
reference frameworkPMCID: PMC5429012
Managing Your Compute Environment
One of the most challenging aspects of bioinformatics workflows is reproducibility. In addition to documenting your analysis with a notebook, providing a copy of your compute environment limits variability in results, allowing for future reproduction of results. A world of options exist to handle this, although some of the most common options are presented.
AWS Elastic Cloud Computing is a useful service for creating entire virtual machines that can easily be copied and distributed. This option does require a paid account with Amazon, and the costs of storing the images and running instances may add up over time, especially if every analysis is stored in a separate image. Additionally, this option does not isolate the analysis environment from the system environment, potentially leading to changes in analysis output as system libraries are updated over time. The RNA-seq wiki makes heavy use of AWS as a distribution platform.
VirtualBox is a general-purpose full virtualizer that allows you to emulate a computer, complete with virtual disks, a virtual operating system, and any data and applications stored therein. It has the advantage of creating machines that are stored and run on local hardware (e.g. your personal workstation), but the extra overhead of running a virtual computer on top of a host operating system can considerably slow performance of tools stored on the virtual machine, and thus is best used for testing or demonstration purposes.
Docker packages apps and their dependencies into containers which may be docked to a docker engine running on a computer. Docker engines are available on all major operating systems, and allow software to remain infrastructure independent while sharing a filespace and system resources with other docked containers. This is a much more efficient approach than guest virtual machines, and containers may be docked locally or on cloud-based infrastructure.
Conda is a language-agnostic package, dependency and environment management system. It is included in the data-science-focused distribution of Conda, Anaconda. Anaconda is based on Python and R packages for the analysis of scientific, large-scale data. Bioinformaticians also commonly use Bioconda, which add channels to Conda with bioinformatics tools (such as the popular sequence alignment tool BWA).